Pain Physiology
This web-page assumes a basic knowledge of medicine. We assume that the reader is
familiar with such basic concepts as interneurones, synapses, oncogenes, G proteins and
prostaglandins, and has had previous sketchy exposure to neuroanatomy. We will try to
briefly describe the neuroanatomy of pain. Once we have described the main structures
transmitting pain, we will explore the connections between them, neurotransmitters mediating pain, and what happens within
nerve cells carrying painful stimuli. We conclude with a short note on pain pharmacology.
What is pain?
We attach different attributes to pain. If a child injures herself, she will cry, and
say "It's sore". Her mother might enquire "Where does it hurt, darling?" Think of these as two
different approaches to pain - the "motivational-affective" component which is
phylogenetically primitive, and concerned with pain as something nasty to be avoided, and
the evolutionarily more recently acquired "sensory/discriminative" ability to
perceive exactly where the pain is, and respond appropriately. As we explore the
complex structures that mediate our appreciation of and response to pain, we will find
that we can conveniently categorize them into two groups, those that deal with the
response to pain as an unpleasant sensation, and those that
are more concerned with sensory/discriminative aspects of
pain. We will consistently colour code
the pathways concerned with these two 'types' of pain.
Pain in the Cortex
It used to be said that cortical structures are only tangentially involved in the
perception of pain, if at all. This is clearly extremely silly, as a host of connections
link higher cortical structures with pain-centred nuclei in the thalamus and brainstem.
Major cortical players are:
- The primary sensory cortex, S I
- The secondary sensory cortex, S II
- The anterior part of the insula
- The cingulate gyrus.
The next three pictures clearly show their location - S I is concerned with localisation of pain, while the other three structures are thought
to be concerned with the motivational-affective aspects which from now on for the sake of
brevity we will call "affective" pain, although
"sore" pain might be a better term!
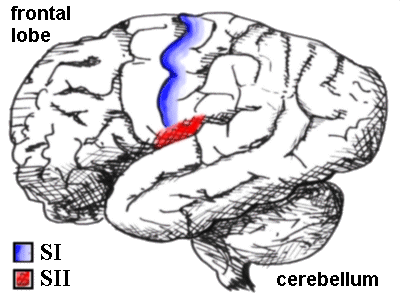
The human brain seen from the side, showing the SI and SII sensory cortex. SI is a
thin strip made up of Brodman areas 3,1 and 2 posterior to the central sulcus, while SII
lurks just above the lateral sulcus.
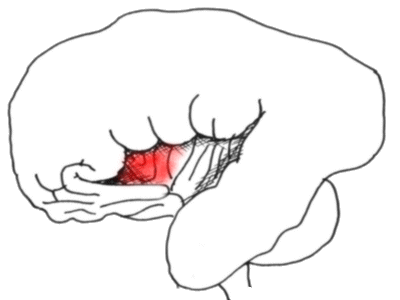
The temporal lobe has been retracted to reveal the insula in all its splendour
- the anterior portion is probably concerned with pain perception.
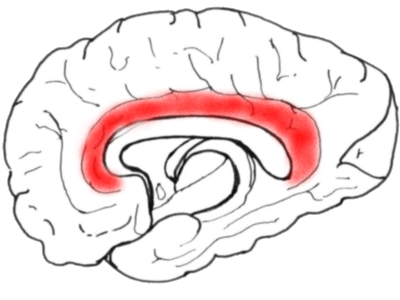
Next we look at the medial aspect of a hemisphere, showing the cingulate gyrus.
The anterior part of the cingulate gyrus is important in the perception of 'affective'
pain.
The Thalamus
The thalamus is the 'central switching station' of the brain. Several of its multiple
nuclei are concerned with pain. The lateral nuclei deal mainly with sensory/discriminative
aspects, the medial ones with 'affective' pain. Because the thalamus is such a complex
three-dimensional structure it's extremely difficult to visualise it. We therefore adopt
the approach of "building it" from medial to lateral (successively adding
nuclei) in the following diagram (follow the arrows):
| Medially we add the poorly-defined midline nuclei.
Lateral to these we plonk on the dorsomedial nucleus (dm) and, more anteriorly the
anterior nuclei (ant). The internal medullary lamina bounds the dorsomedial nucleus
laterally, and separates it from the anterior nuclei. Within the internal medullary lamina
are the intralaminar nuclei, including the centromedian (cm) and centrolateral
nuclei.
Lateral to the internal medullary lamina (iml) lies the ventral posteromedial nucleus
(vpm) and anterior to this is the ventral anterior nucleus (va).
Even further laterally we have the lateral dorsal(ld), lateral posterior(lp), ventral
lateral(vl) and ventral posterolateral nuclei. The pulvinar
(grey) is situated posteriorly. |
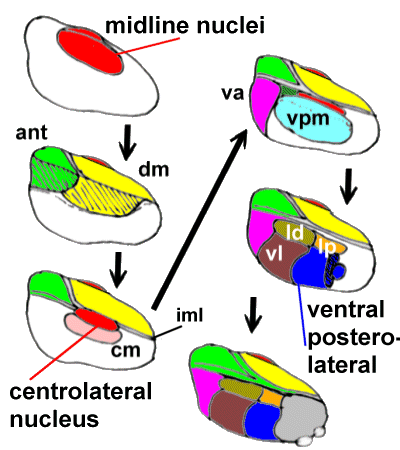 |
A Diagram of the Thalamic nuclei. The lateral thalamus is thought to be mainly concerned
with 'discriminative' pain, the medial with "affect/motivation".
|
Take note of the inconspicuous midline nuclei - the first
group that we put in place as we 'built' the thalamus. They include the:
- nucleus reuniens
- rhomboidal nucleus
- submedius nucleus
Although difficult to identify, and glossed over in most neuroanatomy texts, these
nuclei, particularly the submedius, may turn out to be very important in pain perception!
Midbrain
There is a host of pain-related structures in the midbrain. Most of this circuitry is
involved in 'affective' pain, with extensive connections to
the reticular system of the brainstem. Important components are:
- The peri-aqueductal grey matter (PAG)
- Deep layers of the superior colliculus
- The red nucleus
- The pre-tectal nuclei (anterior and posterior)
- The nucleus of Darkschewitsch
- the interstitial nucleus of Cajal
- The intercolliculus nucleus, nucleus cuneiformis and even the Edinger-Westphal nucleus.
Many of these are shown in the following diagrams:
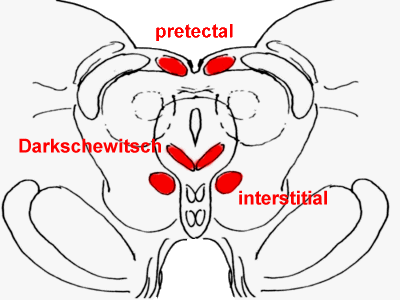
Rostral midbrain, with the anterior pretectal
nucleus, interstitial nucleus of Cajal, and nucleus of Darkschewitsch in red.
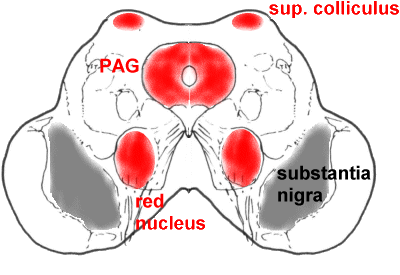
A more caudal section through the midbrain, showing the superior colliculus (SC),
periaqueductal grey matter (PAG), and red nucleus (RN).
The Pons
The most important pain-related nucleus in the pons is probably the locus coeruleus.
This is jam-packed with noradrenaline-containing neurones, and projects to a variety of
brainstem structures that modulate pain through pathways that descend to the spinal cord.
Another notable group is the parabrachial nuclei, which receive a vast number of ascending
spinoreticular fibres. Both are shown in the following cross-section through the pons.
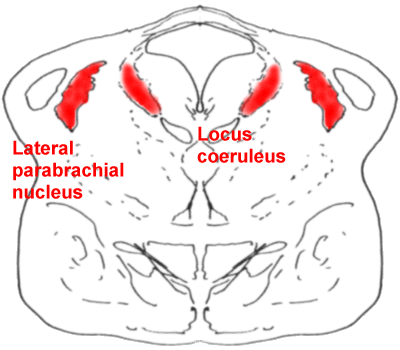
A cross-section through the pons,
showing the locus coeruleus (LC) and the lateral parabrachial nucleus (PB)
The Medulla
This too is involved in the motivational/affective aspects of pain. Important cell
groups are the nucleus gigantocellularis and related nuclei, the lateral reticular
nucleus, and a variety of other nuclei. Some of these are shown in the following diagram.
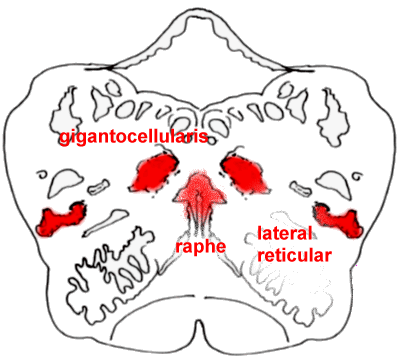
The rostral medulla, with
the nucleus gigantocellularis and lateral reticular nucleus clearly shown.
Take note of the raphe nuclei near the midline, which are important in the descending
pathways that suppress pain.
How do these nuclei tie together?
Connections between the above structures are complex and confusing, but can be divided
into several groups. We give you four:
- 'Discriminative' fibres ascending from the spinal cord to
the lateral thalamus and thence to S I These have been called the neospinothalamic
fibres, because they appeared on the scene long after the:
- 'Affective' fibres that follow a similar course from the
spinal cord, but end up in the reticular formation of the hindbrain. These spino-reticulodiencephalic
fibres have extensive connections throughout the brainstem, and from there project to the
medial thalamus and the cortex (S II).
- Descending fibres that pass down from brainstem to spinal cord, inhibiting
incoming sensations of pain. A lot of these descending fibres originate in the locus
coeruleus, others in the raphe nuclei.
- A variety of interesting and sometimes clinically important connections, some of them
only recently elucidated!
Before we look at the connections in more detail, let's look at pain connections in the
spinal cord:
The Spinal Cord
Traditionally it was thought that most pain fibres entered the dorsal root of the
spinal cord (the "sensory" root) and then synapsed in the dorsal part of the
spinal grey matter, before passing the message up through the spinothalamic tract. It is
now known that this is a gross oversimplification. In fact, anything up to 40% of sensory
fibres enter in the ventral root!
Histologically the grey matter of the spinal cord is divided into ten 'laminae'. The
dorsal part is divided into five laminae (I to V), components of which deal with most
incoming pain fibres. VII is in between these laminae and the more ventral laminae VIII
and IX, and X refers to the grey matter around the central canal of the spinal cord. And
what about lamina VI? This is only discernible in the bulges in the cord related to where
the innervation of the limbs originates.
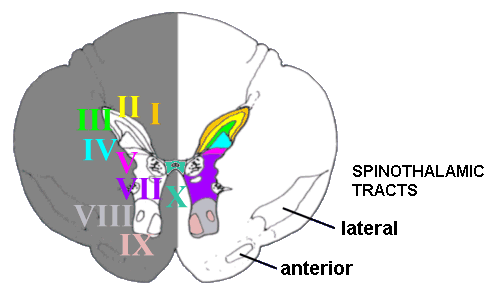
A transverse section through the thoracic
spinal cord, showing the grey matter and various laminae.
Much enthusiasm was generated when the "gate control" theory of pain was
first described. Although this mechanism has now been well documented, and has some
clinical utility, it is known to be a gross over-simplification. The basic idea is that
incoming pain stimuli can be "gated" (shut off) by other stimuli, because many
nerve cells talk to one another in the dorsal horn. Important fibres coming from the
periphery into the dorsal horn include:
- Tiny unmyelinated 'C' fibres that are important carriers of
the long-lasting burning pain that makes a surgical wound (for example) such an unpleasant
experience. There is controversy about where these fibres terminate -
according to Willis and Westlund, in primates many terminate in the deep dorsal horn
and even the ventral horn, although conventionally lamina II has been
said to be their destination.
- Thin myelinated 'A delta' fibres, concerned with more
accurate localisation of pain, and terminating mostly laterally in laminae I and V.
- Rather chunky 'A beta' fibres that carry information about vibration and position sense
from the periphery to the cord.
Unpleasant stimuli entering via the C fibres can be
suppressed by concurrent stimulation of A delta fibres (high
amplitude low frequency stimulation, for example by acupuncture) or by impulses passing
through A beta fibres. Examples of the latter include TENS (transcutaneous electrical
nerve stimulation) and the simple expedient of rubbing the skin, which is well known by
mothers to decrease perception of pain!
Ascending Pain Connections
We now have sufficient resources to examine the various ascending pain pathways. First,
the primitive spino-reticulo-diencephalic connections:
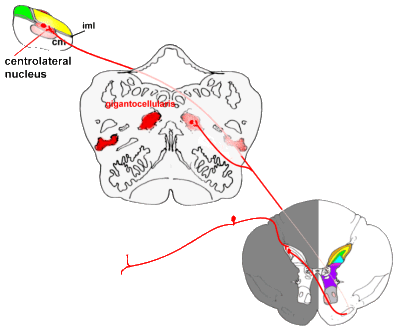 |
Pathways from the spinal cord to the brainstem, and from there to
the thalamus (diencephalon).
Some fibres pass directly to the medial thalamus, while others end in (or send collaterals
to) a variety of nuclei in the brainstem. Connections between the reticular system and
thalamus are not shown for reasons of clarity. |
Next, we examine the phylogenetically more modern pathways from cord to lateral
thalamus and thence to the S I cortex. These pathways are discriminative pain
pathways, and have little to do with perception of pain as a 'sore' stimulus! These
pathways have few or no opioid receptors - morphine (for example) will have no effect on
such pathways!
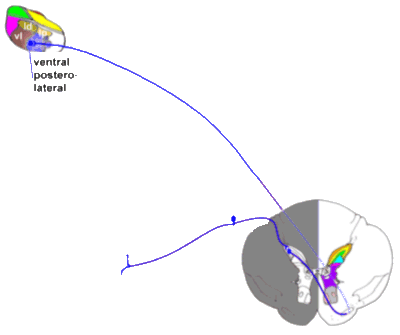 |
The neospinothalamic tract.
|
Descending Pain Connections
As important as the ascending pathways are fibres that descend from brainstem to spinal
cord to modulate the incoming signals. Notable neurotransmitters mediating this
anti-nociceptive effect include noradrenaline (norepinephrine), especially in the locus
coeruleus, and serotonin in the raphe nuclei. Opioid receptors are prevalent here. Some
descending connections are:
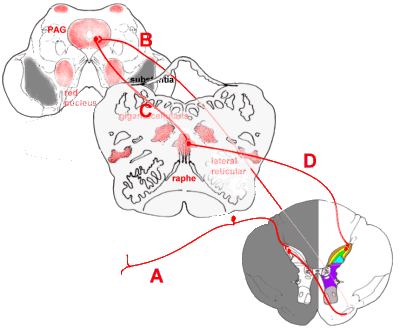 |
Descending connections that modulate incoming pain impulses.
Incoming painful stimuli are transmitted (A) to the dorsal horn, and from there (B) to the
periaqueductal grey (PAG). Descending impulses pass (C) to the raphe nuclei, especially
the nucleus raphe magnus, in the upper medulla, and thence back to the dorsal horn via
reticulospinal fibres (D). The above shows only the serotonergic descending fibres.
Other pain-suppressing impulses pass from the PAG to the locus coeruleus, and from there
to the dorsal horn. |
Pain in the periphery - the nociceptor
The above connections are awfully complex. One might think that once we moved out to
the periphery, things might become more simple. Not so! Most tissues are well provided
with specific pain receptors called nociceptors. Formerly it was thought that
painful stimuli were detected through 'overstimulation' of receptors for other modalities.
This is incorrect. The quality of the pain perceived on stimulation of nociceptors seems
to depend on the site of stimulation, and the nature of the fibres transmitting the
sensation. Even in the periphery, there is a distinction between the sharp immediate pain
("first pain") transmitted by A
delta fibres, and the prolonged unpleasant burning pain
mediated through the smaller unmyelinated C fibres.
Nociceptors have numerous different receptors on their surfaces that modulate their
sensitivity to stimulation. These include GABA, opiate, bradykinin, histamine, serotonin
and capsaicin receptors, but the various roles of these receptors are poorly
characterised.
The most fascinating aspect of pain perception in the periphery is that normally most
nociceptors lie dormant. Inflammation sensitizes this vast population of nociceptors,
making them far more sensitive to stimulation (hyperalgesia). Hyperalgesia may be primary
(felt at the site of stimulation, related to sensitization of the neurones innervating
that area) or secondary (felt at a site remote from the original injury, and probably
related to NMDA-mediated "wind-up").
& Neurotransmitters
A plethora of neurotransmitters mediates transmission of the sensations of pain in both
brain and spinal cord. The list is intimidating, and grows daily. We can try and 'lump'
these neurotransmitters into various groups:
- Excitatory neurotransmitters:
Important are glutamate and the tachykinins, agents that act at the various
neurokinin receptors including as substance P ('P is for pain'), neurokinin A and
neurokinin B. Other substances that transmit pain impulses from incoming nerves in the
dorsal horn including calcitonin gene-related peptide, vasoactive intestinal polypeptide,
somatostatin and bombesin.
- Inhibitory neurotransmitters:
There are several inhibitory neurotransmitters, but in the central nervous system, gamma
amino butyric acid (GABA) appears to reign supreme. Over forty percent of
inhibition in the mammalian central nervous system is GABAergic.
- Neurotransmitters involved in Descending Pain Regulation:
Here, the alpha-2 stimulatory effects of noradrenaline (norepinephrine) and the
effects of serotonin are prominent. Opiates relieve pain by stimulating mu
and delta receptors at a host of sites.
Specific neurotransmitters
Glutamate
A brief Medline search for articles using the abbreviation "NMDA" in the past
ten years will garner about twelve thousand references. This attests the fanatical
interest researchers have in this, the hottest of the glutamate receptors, but one mustn't
forget that there are at least two others, the "AMPA" receptor and the obscure
and devious metabotropic receptor. The NMDA receptor mediates a host of spinal responses
to severe painful stimulation, but there are several catches to understanding how it
works. Normally, the receptor is inactive as it is physiologically choked by a magnesium
ion sitting in its ion channel. In order for this ion to be removed, adjacent peptide
receptors have to be stimulated - the Mg++ then pops off, and an emphatic
painful response occurs. Neurophysiologists have known about this phenomenon for ages,
gracing it with the label "wind-up" - as the frequency of C-fibre stimulation
increases there is a dramatic and long-lasting central response, with some populations of
spinal neurones becoming more and more sensitive to stimulation.
Consequences of glutamate receptor activation include production of c-fos (discussed below) and spinal production of prostanoids and the ubiquitous Dr NO,
nitric oxide. Unfortunately all this knowledge benefits clinicians surprisingly little, as
drugs that antagonise the effect of glutamate at the NMDA receptor tend to induce
psychosis in humans, but the combination of low dose NMDA antagonists with opioids may be
supra-additive with fewer side effects.
GABA
GABA is widespread in the brain and spinal cord. Together with its partner glycine, it has
major inhibitory effects, dramatically evident in poisoning with strychnine, which
antagonises glycine. Interneurones in laminae I, II and III are GABA-rich, and mediate
gate control in the dorsal horn by synapsing on neurones that contain substance P.
There are several distinct GABA receptors that work quite differently - the GABAA
receptor is a "ligand-gated ion channel" that allows chloride ions to leak into
the cell, while the GABAB receptor is a "seven-spanning"
transmembrane structure that activates G proteins.
Again, the clinical utility of this knowledge is small, as GABAB receptor
agonists such as baclofen, which are analgesic at the spinal level in rats, have little
effect in man, although they may potentiate the analgesia of morphine. Benzodiazepines
modulate the GABAA receptor allosterically - but GABAA seems more
important at supraspinal than spinal sites.
Tachykinins
It will probably be several years before newer agents such as neurokinin antagonists have
been tested sufficiently for widespread clinical use, although (for example) NK-1
antagonists such as CP-96 345 have been shown to moderately decrease isoflurane MAC in
tail-clamped rats (shudder!) when given intrathecally. Neurokinin receptors probably do
mediate pain in the spinal cord - substance P binds to the NK-1 receptor while neurokinins
A and B bind respectively to the NK-2 and NK-3 receptors. Collectively these substances
are known as 'tachykinins'. The tachykinin receptors are G-protein coupled, and increase
intracellular calcium levels, triggering gene transcription.
at the Cellular Level
Probably the most significant discovery ever in the field of pain has been the
gene c-fos. The cellular analogue of a viral oncogene, this rather special gene and its
cellular product, the protein called Fos seem crucial to the profound central
nervous system changes that occur when an animal (or man) feels pain. Central nervous
system c-fos expression correlates extremely well with painful stimulation. Generically,
Fos is one of the inducible transcription factors (ITFs) that controls mammalian
gene expression.
We now have a molecular marker for pain! Even more important, we know that
because c-fos is a proto-oncogene - that is, it can promote vast intracellular changes
including cellular restructuring and proliferation - it is almost certainly involved in
the long-term neurological consequences of noxious stimulation.
Noxious peripheral stimulation not only causes Fos to appear in the spinal cord, but
also the ITFs called Jun and Krox. Certain "constitutive transcription factors"
also change their activity (for example, CREB becomes phosphorylated). Brief stimulation
(10 min) causes ITFs to appear within 30 min, peak at one to two hours, and disappear
within about eight hours. Prolonged stimulation causes a many-fold increase in ITF
expression, and substantially prolongs expression. Nociceptive C-fibre stimulation seems
to be the main stimulus for ITF production in the spinal cord, as is the case with painful
visceral stimulation (for example, introduction of cyclophosphamide into the bladder). In
the latter c-fos appears within an hour in laminae I and II, the parasympathetic column,
the dorsal grey commissure, as well as in the hindbrain (periaqueductal grey matter, locus
coeruleus, the parabrachial area, and several other areas including the dorsal vagal
complex and ventrocaudal bulbar reticular area).
Centrally, the reuniens, rhomboid and submedius nuclei all express c-fos, as do a
number of areas in the cortex, thalamus, hypothalamus and amygdala. More caudally, the
parabrachial nucleus also lights up!
A remarkable finding is that with prolonged stimulation, c-fos disappears from spinal
neurones after two to seven days! This disappearance is despite increased neuronal
excitability and a marked increase in expression of neurokinin and glutamate receptors,
and may be simply because the neuronal changes are fixed, so the ITF is no longer needed!
Conversely, chronic lesions of sensory nerves (for example, the neurophysiologist's
favourite, partial sciatic nerve ligation) can induce chronic c-fos expression even in
nerves which don't normally express the ITF!
Consequences of ITF production include neuropeptide production and synthesis of a
variety of receptors. Fos is involved in cell replication and differentiation, but we are
still horribly confused about its precise effects on neurones.
Fos may be important in expression of genes for
dynorphin, enkephalin and tyrosine hydroxylase, for example, as these all have binding
sites for AP1, the transcription complex that actually binds to DNA and does the job! (AP1
is the term used to describe the combination of Fos and the ITF Jun, or the dimer Jun:Jun.
AP1 seems to bind the DNA consensus sequence TGACTCA).
It is now well known that "anaesthesia" (for example the combination of
halothane and N2O) does not suppress production of c-fos within the spinal cord,
despite an apparent "adequate" level of anaesthesia. Fentanyl reduces c-fos
production by about 50%, and appropriate neuraxial block with local anaesthetic agents can
almost totally ablate the c-fos response.
Opiates
Opioids act by stimulating mu, delta and kappa receptors. Other receptors (such as
sigma and epsilon) have been postulated, but probably don't exist. The status of opioid
receptor subtypes (mu1, mu2, delta 1, delta 2 and so on) is also controversial. If one
looks at the genes for these receptors, there do not appear to be subtypes, but this
doesn't mean that post-translational modification may not have created distinct subclasses
of, say, the mu receptor. Of more interest is the identification of a so-called
"orphan" receptor called ORL-1 (for "opiate-like receptor 1"), with
about 60% sequence homology with the other opiate receptors. An endogenous ligand that
acts at ORL-1 has recently been identified and called nociceptin, otherwise known as
orphanin F2.
Even more recently its precursor, prenociceptin, has been
found to give rise to other opioid-like substances called Noc II, Noc III and nocistatin,
but the function (if any) of all these fancy new substances is unclear, to say the least!
A variety of G proteins are associated with opioid receptors, including Gi
and Go. Generally, mu and delta agonists seem to decrease cyclic AMP
production, and hyperpolarise membranes by stimulating an inward rectifying potassium
channel. This hyperpolarisation decreases the release of neurotransmitters from the nerve
cell, as such release depends on opening of voltage-sensitive calcium channels.
Many researchers have tried to find out where opiates work in the central nervous
system. The best-characterised site is undoubtedly the periaqueductal grey, where morphine
micro-injection is profoundly analgesic. Other probable sites of action include the
mesencephalic reticular formation, the substantia nigra, and the nucleus gigantocellularis
& areas near the nucleus raphe in the medulla. Mu receptors also seem to be present in
the amygdala, and the ventral forebrain.
Descending control mechanisms: Adrenergic and serotonergic pathways from the
brainstem to the spinal cord inhibit incoming painful stimuli. Opiates release these
pathways from GABAergic inhibition, increasing activity in the descending pathways and
thus suppressing pain. Stimuli for opiate release in for example the periaqueductal grey
include pain stimuli ascending from the spinal cord, and stimuli passing along the
multiple connections that the brainstem pain structures have with other local nuclei, the
midbrain, and higher centres.
Spinal opioid receptors: These are prevalent and are very effective in
modulating pain - in lamina I the predominant receptors seem to be mu and delta, and in
laminae II to V, kappa.
Peripheral opioid receptors: Although some studies have shown an inhibitory
effect of opiates on peripheral nociceptors, the mechanisms and relevance of such
observations remain uncertain. If these peripheral opioid receptors (predominantly mu and
kappa) have a function, it is probably mainly in the terminals of C fibres already
sensitized by inflammation. Practically, intra-articular morphine has a powerful
analgesic-sparing effect after knee surgery! Another unusual revelation is that immune
cells appear to be profoundly influenced by opiates, following stimulation of their delta
and kappa receptors.
Non-steroidal anti-inflammatories (NSAIDs)
Classically the effectiveness of NSAIDs against inflammation has been attributed to
their effects in inhibiting prostaglanding biosynthesis. They do this by several different
effects on cyclo-oxygenase (COX), the crucial enzyme in the initial synthesis of
prostaglandins:
- irreversible inactivation of COX; or
- reversible competitive inhibition; or
- reversible non-competitive inhibition ("free-radical trapping")
Aspirin, the prototype NSAID, acts through the first mechanism, irreversibly
acetylating COX. Many drugs such as ibuprofen, mefenamic acid and sulindac are reversible
competitive inhibitors, and (surprisingly) paracetamol may scavenge hydroperoxides
generated during arachidonic metabolism, and thus quench prostaglandin formation (The
hydroperoxides normally stimulate COX, causing positive feedback)!
Much in the news has been the identification of two subtypes of COX. COX-1 is the
constitutive variant that makes prostaglandins vital for protecting the stomach through
mucus production, and maintenance of renal blood flow. COX-2 is the inducible form that
mediates the pain of inflammation by sensitising peripheral nociceptors. Most conventional
NSAIDs inhibit COX-1 more than COX-2, consequently pharmaceutical companies have invested
fortunes in making selective COX-2 inhibitors.
NSAIDs have many other effects far removed from their COX inhibition. These include
cell membrane effects (inhibition of neutrophil aggregation, inhibition of H2O2
generation (piroxicam), and even phosphodiesterase inhibition (indomethacin, diclofenac).
Some of these effects may be mediated through interference with G protein function. In
addition, NSAIDs may have central antinociceptive effects! This has been seen both
experimentally (NSAIDs prevent the usual rise in cerebrospinal fluid prostaglandins after
NMDA receptor activation) and clinically. Descending serotonergic pathways seem to be
activated by NSAIDS, and part of the central action of NSAIDs in animal models appears to
be prevented by naloxone! In addition, NSAIDs may interfere with spinal production of
nitric oxide in response to NMDA receptor stimulation. They may even reduce c-fos
expression!
Summary
In confronting the bewildering complexity of pain pathways, we tend to forget that
fundamentally, above all else, pain is sore! There is
another aspect of pain, that is, the localisation of a painful stimulus, but long before
our primitive ancestors acquired the high-tech pathways necessary to perceive the sensory-discriminative aspects of pain, they were still sore. Throughout the nervous system, we can identify these two
forms of pain transmission. In the periphery we have fast pricking
pain transmitted by myelinated A-delta fibres, and 'slow' burning pain mediated by the primitive C
fibres. In the spinal cord, the affective component of pain is transferred by
spinoreticular fibres, and fibres to the medial thalamus, and thence to those parts of the
cortex where 'sore' pain is perceived. The phylogenetically
newer 'neospinothalamic tract' transmits discriminative components of pain to the SI
cortex via the lateral thalamus.
As important as the ascending pathways, are descending pathways that modulate the
incoming signal. These are predominantly noradrenergic and serotonergic, and can be
modulated by stimulation of opiate receptors. Opiates, still the mainstay of therapy for
moderate to severe pain, have a variety of receptors that are widespread in the central
nervous system. Only now are we beginning to understand the subcellular mechanisms of
pain, particularly how c-fos works. We hope that such understanding will eventually help
us to manage more effectively man's second greatest scourge - pain.
References
From the vast pain literature, we've pulled out a few good articles and books for your
delectation. We used these extensively in constructing the above review.
Brilliant Reviews
- Cashan JN. Drugs 1996 52 S5 13-23. The Mechanisms of Action of NSAIDs in Analgesia. An
incredibly well-written article.
- Cross SA. Mayo Clin Prog 1994 69 375-83. Pathophysiology of Pain. Still one of the
best reviews I've come across.
Worthwhile Reading
- Willis WD & Westlund KN. J Clin Neurophys 1997 14 2-31. Neuroanatomy of the Pain
System and of the Pathways that Modulate Pain. A comprehensive and recent review, not
for the faint-hearted
- Herdegen T & Leah JD. Brain Research Reviews 1998 28 370-490. Inducible and
constitutive transcription factors in the mammalian nervous system: control of gene
expression by Jun, Fos and Krox, and CREB/ATF proteins. A mammoth review of c-fos and
other transcription factors
- Malcangio M & Bowery NG. TiPS 1996 17 457-62. GABA and its receptors in the spinal
cord. A concise, worthwhile review of gamma amino butyric acid.
- Yaksh TL. Acta Anaes Scand 1997 41 94-111. Pharmacology and mechanisms of opioid
analgesic activity. A good review of opiates.
- Dickenson AH. Acta Anaes Scand 1997 41 112-115. NMDA receptor antagonists: interactions
with opioids. Brief.
- Carpenter MB, Sutin J. Human Neuroanatomy. 8ed. 1983. Pick up a more recent edition
if you can!
A few Web References on Brain Anatomy!
